







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
稻秸蓝藻沼气发酵过程中微生物群落结构解析
[刘爱民 , 曹圆圆, 刘宇, 董德武, 卢存龙]
, 曹圆圆, 刘宇, 董德武, 卢存龙]
, 曹圆圆, 刘宇, 董德武, 卢存龙]
|
|


作者简介:刘爱民(1968—),女,安徽太和人,副教授,博士,研究方向为应用微生物,E-mail:amliu9393@163.com。
采用自行设计的可控恒温厌氧发酵装置,以水稻秸秆和铜绿微囊藻为发酵原料,分别对稻秸进行生物(平菇发酵液、沼渣)和化学(6% NaOH)方法预处理,在(35±1)℃的条件下进行厌氧发酵。每隔5 d,采取不同处理组的发酵液,提取DNA,进行PCR扩增,通过高通量测序技术分析不同处理组样品的微生物群落组成、丰度和多样性。结果显示,各处理组的微生物群落结构有所差异,在门水平上,以变形菌门、拟杆菌门和厚壁菌门为主,但仅平菇处理组的沼液中含有梭杆菌门。此外,平菇处理组的Shannon指数亦高于其他2组,具有较高的菌群丰度和多样性。平菇处理组的稻秸产气性能最稳定,可能与其微生物群落多样性丰度最高有很大关系。
我国是农业大国, 每年农作物秸秆的产量约7亿t[1], 主要为粮食作物秸秆、油料作物秸秆、棉花秸秆和麻类作物秸秆等。然而, 我国秸秆的利用率极其低下, 大部分自然腐烂或就地焚烧, 不仅浪费资源, 而且污染环境, 由此造成的直接损失难以估计[2]。如果将这些“ 废弃物” 很好地加以利用, 如利用厌氧消化技术将稻草秸秆转化为清洁、高效的能源— — 沼气, 不但能在一定程度上解决农村能源紧缺的问题, 而且可以改善农村生态环境[3, 4]。
有机物厌氧发酵产沼气主要分为水解发酵、产氢产乙酸以及产甲烷3个阶段, 每个阶段都有不同的微生物发挥作用。其中:水解发酵与产氢产乙酸阶段主要由细菌参与, 涉及一系列复杂的化学代谢过程, 生成乙酸等挥发性有机酸; 产甲烷阶段主要由产甲烷古菌参与, 将乙酸转化为甲烷、氢气和二氧化碳等[5]。秸秆的厌氧降解需要多种类型的微生物协同作用, 但是目前人们对沼气发酵微生物, 尤其是厌氧微生物的生理生化特性及代谢特点的认知程度极其有限[6], 导致发酵启动周期长、底物降解率低和运行不稳定, 已成为秸秆发酵技术发展的瓶颈。尽早明确秸秆发酵过程的微生物群落结构与功能, 对于提高秸秆的降解率及增加产气量具有重要的指导意义[7, 8]。
现有的微生物群落结构研究方法主要包括分离培养和分子生物学方法。传统的分离培养不但操作烦琐, 费时费力, 更重要的是环境中微生物群落结构非常复杂, 且自然界存在大量不可培养的微生物, 导致该方法并不能全面客观地反映环境中微生物群落结构及其功能。随着分子遗传学和分子生物学技术的飞速发展, 许多新的生物学手段可被用于分析微生物群落结构, 常用的有:随机扩增多态性DNA分析(RADP), 限制性片段长度多态性技术(RFLP), 荧光原位杂交技术(FISH), 变性梯度凝胶电泳技术(DGGE)和温度梯度凝胶电泳技术(TGGE)等。这些技术以复杂的环境样品为研究对象, 可以不经培养, 直接提取其中微生物的基因组DNA, 利用分子生物学手段进行分离、鉴定和定量, 克服了传统的纯培养技术的缺陷, 直接从分子水平反映厌氧发酵过程中微生物种群的多样性和结构组成; 但是, 这些分子生物学方法普遍具有通量低、分辨率低和成本高等局限性, 仍不能完全反映微生物群落的整体信息[9]。
高通量测序技术不仅可以对数百万个DNA分子同时测序, 而且可以同时对上百个样本进行测序分析, 是解析复杂环境中微生物群落组成和相对丰度的重要工具。与传统的分子生物学技术相比, 新一代高通量测序技术具有高通量、低成本、可以同时对多个单独的DNA分子进行测序分析等优点[10]。目前, 高通量测序技术已经被广泛应用于不同自然环境条件下(如海洋、湖泊、土壤、垃圾填埋场以及人和动物的肠道等)微生物群落结构和功能的研究[11, 12, 13]。据此, 本研究拟应用高通量测序技术分析稻草秸秆和蓝藻沼气发酵过程中的微生物群落结构和多样性, 以期为秸秆沼气发酵工程应用提供理论参考。
1.1.1 发酵原料
试验材料为稻草秸秆、铜绿微囊藻和活性污泥。稻草秸秆采自安徽省芜湖市田间, 剪短至2 cm以下待用; 铜绿微囊藻(Microcystis aeruginosa FACHB-942)来自中国科学院武汉水生生物研究所; 活性污泥采自芜湖市某啤酒厂的污水净化处理池。
1.1.2 稻秸预处理
试验所用秸秆共设3种预处理:1)沼渣处理, 将经过稻草秸秆和蓝藻厌氧共发酵30 d后的沼渣用清水冲洗干净, 除去里面的污泥, 保留未完全降解的稻草秸秆, 晾干备用。称取上述发酵过的稻草秸秆300 g, 备用; 2)平菇发酵液处理, 从活化好的平菇斜面培养基上取一块5 mm2的菌块, 接入PDA液体培养基中[14], 28.5 ℃, 170 r· min-1培养3 d, 按10%的接种量再转接到新鲜PDA液体培养基中, 同上条件培养7 d备用。称取300 g稻草秸秆装入5 L发酵罐中, 接种上述已培养好的平菇种子液1.5 L混匀, 28.5 ℃恒温培养7 d后晾干备用; 3)NaOH处理, 称取300 g稻草秸秆装入5 L发酵罐中, 用6%的NaOH溶液处理, 28 ℃恒温浸泡3 d, 并水洗除去碱液, 晾干备用。
1.1.3 铜绿微囊藻培养和处理
将铜绿微囊藻藻种以1:1的比例接入BG11培养基中[15], 置于光照培养箱中培养。培养条件:温度25 ℃, 光照强度2 000 lx, 光暗周期12 h:12 h。每天早中晚各摇瓶1次, 以防止藻细胞黏附于瓶壁上[16, 17]。将培养好的藻液离心, 除去90%(V/V)上清后待用。
1.1.4 活性污泥驯化
将活性污泥取回后厌氧保存在塑料容器中, 加入适量的葡萄糖稀溶液, 实验室驯化7 d。驯化后的污泥质地蓬松, 呈絮状, 颜色为灰黑色, 并夹带少量气泡, 测定pH值7.0。
本试验所使用的厌氧发酵罐为自行设计、工厂代加工器材, 最大容积5 L, 发酵容积4 L。罐的内层进行发酵, 外层连通循环水保温; 发酵罐的顶部有2个出气孔, 通过阀门控制以便放出沼气, 通过橡胶管连通集气瓶, 采用排水法收集沼气; 发酵罐的外层侧部有2个阀门控制循环水的进出, 通过循环水的畅通保证整个发酵过程在(35± 1)℃的温度下进行。整套厌氧发酵装置如图1所示。
 | 图1 厌氧发酵罐的装置 |
本试验主要研究铜绿微囊藻协同秸秆处理下沼气发酵过程中的微生物群落结构和多样性, 以及沼气发酵状况。将经沼渣处理、NaOH处理和平菇发酵液预处理过的秸秆, 分别连同100 mL铜绿微囊藻浓缩液及1.5 L活性污泥加入5 L的厌氧发酵罐中, 混合均匀后, 调pH值至7.0, 迅速封盖, (35± 1)℃恒温水浴条件下发酵, 分别标记为处理1、处理2和处理3, 每处理重复3次。
采用水压式法收集发酵产生的气体, 根据排出饱和食盐水的体积计算每日产生气体的体积。从进行发酵的第1天开始, 每天定时记录各套装置的产气量, 以3个平行样品的平均产气量作为最终产气量。
1.5.1 样品采集和总DNA提取
从进行装罐发酵的第1天开始, 每5 d量取3 mL的发酵液样品置于50 mL无菌的离心管中, 加入13.5 mL CTAB提取缓冲液(100 mmol· L-1 Tris, 100 mmol· L-1 EDTA, 100 mmol· L-1 Na3PO4, 1.5 mol· L-1 NaCl, 1.0% CTAB, pH=8.0)和25 μ L蛋白酶K(20 mg· mL-1), 混合均匀后, 37 ℃, 225 r· min-1振荡30 min, 然后加入1.5 mL 20%的SDS, 65 ℃水浴加热2 h。将上述样品处理液3 000 r· min-1离心5 min, 收集上清液, 加入等体积的氯仿轻微振荡混匀, 稍微静置后, 9 000 r· min-1离心5 min取上清液, 加入等体积的预冷的无水乙醇, 4 ℃下过夜, 促进DNA沉淀, 9 000 r· min-1离心5 min, 用TE缓冲液溶解沉淀即为所得的基因组DNA粗提液[18]。在1%的琼脂糖凝胶中电泳检测提取到的DNA质量。
样品及其编号分别为:X1, 处理1第5天样品; X2, 处理1第10天样品; X3, 处理1第20天样品; X4, 处理2第5天样品; X5, 处理2第10天样品; X6, 处理2第20天样品; X7, 处理3第5天样品; X8, 处理3第10天样品; X9, 处理3第20天样品。
1.5.2 16S rRNA基因扩增和高通量测序
样本DNA各50 μ L, 干冰条件下送往南京普东兴生物科技有限公司, 采用紫外分光光度计和琼脂糖凝胶电泳进行浓度及纯度检测。对合格样本进行16S rRNA基因V3区扩增, 通用引物分别为338F(5'-ACTCCTACGGGAGGCAGCAG-3')和518R(5'-ATTACCGCGGCTGCTGG-3'), PCR扩增产物用Ion Torrent Personal Genome Machine平台进行高通量测序。
1.5.3 测序结果质量控制及数据分析
对原始数据首先进行质量控制, 舍弃低质量序列。将获得的序列通过SolexaQA软件去掉平均质量值低于20的序列, 去掉长度小于75 bp的序列, 去掉包含N的序列, 去掉不能完全比上barcode的序列, 并去除barcode和引物序列, 得到有效的序列文件。将相似度大于97%的DNA序列归为1个操作分类单元(operational taxonomic units, OTU), 利用QIIME软件对优质序列进行OTU聚类, 用于样品间的相似性分析, 绘制样品的稀疏曲线图, 评估不同样品的多样性。从每个OTU序列中挑选出具有代表性的序列, 与参考数据库Greengene[19]进行比对, 采用RDP进行分类信息划分, 得到每条序列的分类单元。物种分类单元通常分为6层(界、门、纲、目、科、属), 并对样品的物种分类进行比较, 分析沼气发酵过程的微生物菌群相对丰度。
如图2所示, 各处理组从启动发酵的第1天即开始产气。发酵初期, 产气量逐渐升高, 产气速率较快。处理1在第1天的产气量高达832.5 mL, 明显高于其他处理, 而在第2天产气量出现较大的回落, 在第4天产气趋稳后逐渐上升, 并在第6天达到产气高峰, 之后逐渐回落。这可能是因为秸秆通过一次发酵后仍然有大量的有机物残留在固体残余物沼渣中, 未被微生物降解, 而且沼渣中仍含有较高的纤维素和半纤维素, 是较好的发酵原料, 具有很强的产气潜力[20]。处理2的秸秆产气含量一直保持上升状态, 在第5天达到产气高峰, 为1 170.3 mL, 之后逐渐回落。这可能是由于NaOH中的氢氧根离子破坏了木质纤维结构, 使被木质素包裹的纤维素和半纤维素裸露出来, 而且还破坏了细胞壁结构, 使得厌氧微生物和木质素、纤维素、半纤维素的接触面积大大提高[21]。处理3前2 d产气很平稳, 然后逐渐上升, 在第5天达到产气高峰, 并在这一水平上维持了一段时间后才缓慢下降。
 | 图2 各处理秸秆产气量变化 |
处理1第1天的产气量明显高于其他处理, 可能是因为秸秆已经过一次厌氧发酵, 在发酵微生物的作用下, 残留了很多未被完全利用的有机物质, 能够直接为本次厌氧发酵所利用, 产生大量气体。各处理组在发酵初期产气量逐渐升高, 然后逐渐回落。这是由于在预处理后, 水稻秸秆组分被降解, 能很容易地被接种物中的产酸菌和产甲烷菌迅速利用, 进而产生大量气体, 随着反应的进行, 这些组分被大量消耗而减少, 底物浓度也比开始时小, 所以产气量也逐渐减少。处理3产气时间最长, 累计产气量最高, 就稳定性能来看, 该处理厌氧发酵产气效果最为理想。这可能是因为采用平菇、稻秸和蓝藻混合发酵, 有利于应用不同废弃物中的营养成分和多种微生物的共同作用使发酵过程最优化, 刺激微生物的协同效应, 增加有机负荷从而提高产气量[22]。
2.2.1 16S rDNA扩增
将扩增产物用2%的琼脂糖凝胶电泳检测, 结果显示得到约150 bp的条带(图3)。
 | 图3 16S rDNA PCR扩增结果泳道编号1~9分别为样品号X1~X9, M为分子量标记 |
2.2.2 序列数目和多样性指数
本试验测序样品数9个, 经过对焦磷酸测序所得序列进行质量和长度筛选, 结果得到716 284条高质量的优化序列, 在97%水平上, 一共得到14 490个OTU。
从图4可以看出, 稀疏曲线显示序列较少时, OTU的数目剧增, 随着测序量的不断增大, 曲线最终趋于平缓, 但仍然未达到饱和状态。尽管随着测序量的增大, 新的物种不断出现, 但本次试验的测序量已经基本能够反映样本中微生物多样性的组成。
 | 图4 各样本的稀疏曲线 |
从图5可以看出, 处理3的Shannon指数最大, 高于其他处理, 表明其菌群多样性最高。其次为处理2, 处理1的最低。
 | 图5 各样本的Shannon曲线 |
2.2.3 基于分类地位的群落特征
如图6所示, 在微生物分类门水平上, 共出现了28个已知的细菌门, 占微生物总量的83.1%~88.1%, 未知细菌占11.9%~16.9%。各处理组的样品共同拥有8个优势微生物类群(丰度> 0.8%为优势类群), 分别为变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)、厚壁菌门(Firmicutes)、热袍菌门(Thermotogae)、螺旋体门(Spirochaetes)、互养菌门(Synergistetes)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)。其中, 变形菌门(Proteobacteria)、拟杆菌门(Bacteroidetes)与厚壁菌门(Firmicutes)为主要优势菌群, 这与前人研究结果一致[8, 23]。此外, 处理3的沼气发酵液中独有梭杆菌门(Fusobacteria), 丰度约为0.1%。变形菌门在3组处理沼气发酵罐中相对丰度均最高(19.9%~49.4%), 该门细菌已在厌氧消化污泥、土壤、粪便等环境中发现, 具有水解发酵作用, 可以利用淀粉、长链脂肪酸、氨基酸等, 生成小分子物质, 以供产甲烷菌利用; 拟杆菌门(Bacteroidetes)是第2大优势细菌种群(16.3%~27.4%), 该门细菌已在厌氧消化污泥、废水处理反应器、餐厨垃圾厌氧反应器、肠道等厌氧环境中被发现, 有降解大分子碳水化合物产酸的功能; 厚壁菌门(Firmicutes)是第3大优势种群(7.1%~35.8%), 主要分离自土壤、厌氧活性污泥、人和动物粪便等, 可以利用纤维素、葡萄糖、乳糖等物质, 最终生成丙酸、甲酸和氢等小分子物质。
 | 图6 门水平上各样本细菌群落组成的相对丰度 |
在属水平上进行分类, 可得到70个属(图7)。多度最高的前10属分别为:Thiothrix、Kosmotogae、Treponema、Bacteroides、Clostridium、Phenylobacterium、Rhodobacter、Dok59、Paludibacter、Hyphomonas。其中, Thiothrix是最优势的属, 占总OTU数目的1.0%~4.7%, 属于Proteobacteria门、Gammaproteobacteria纲、Thiotrichales目、Thiotrichaceae科。其次是Kosmotogae, 占0.8%~3.8%, 属于Thermotogae门、Thermotogae纲、Thermotogales目、Thermotogaceae科。Treponema占0.8%~2.2%, 属于Spirochaetes门、Spirochaetes纲、Spirochaetales目、Spirochaetaceae科。Bacteroides和Paludibacter同属于Bacteroidetes门, Clostridium属于Firmicutes门, Phenylobacterium、Rhodobacter、Dok59和Hyphomonas同属于Proteobacteria门。
 | 图7 属水平上各样本中细菌群落组成的相对丰度 |
2.2.4 基于FastUniFrac的群落结构
由图8可看出, 各处理组的样品均比较分散, 可以区分开, 相同处理的样品距离比较近, 说明相同处理的样品细菌群落比较相似, 不同处理的样品细菌群落结构差异较大。
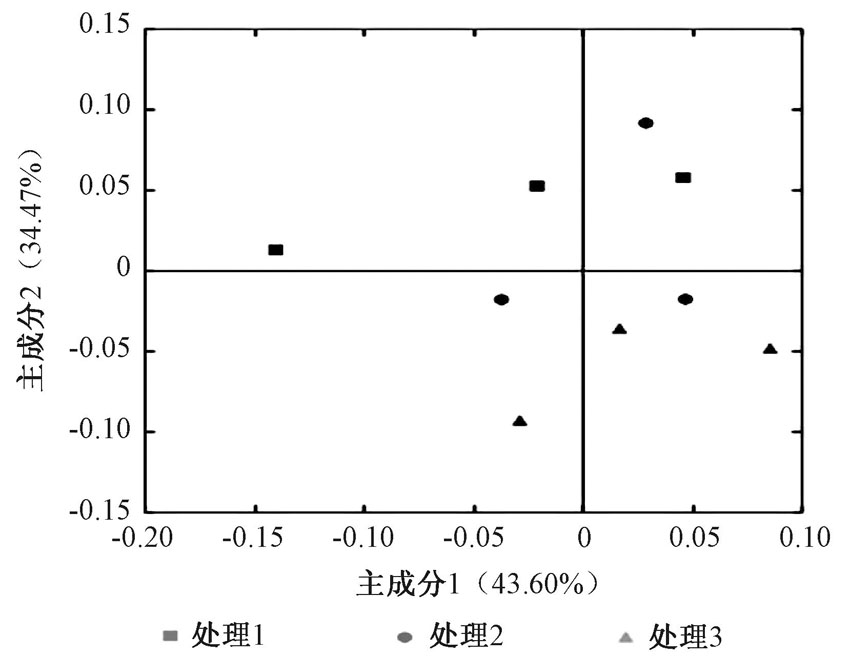 | 图8 基于FastUniFrac的群落结构主成分分析 |
通过对稻秸进行生物(平菇发酵液、沼渣)和化学(6% NaOH)方法预处理, 并对稻秸蓝藻厌氧发酵的沼液样品16S rRNA基因的V3区进行高通量测序, 发现平菇发酵液处理的菌群多样性高于其他处理组, 且产气性能最稳定, 推测可能与其微生物群落多样性丰度最高有很大关系。
The authors have declared that no competing interests exist.
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|